Topic index: A
B C
D E
F G
H I
J K
L M
N O
P R
S T
U V
W X
Z
Procedures Treatments
Home
E
Ecthyma | Eczema
or dermatitis | Elastosis perforans
serpiginosus | Ephelides | Epidermal
naevi | Epidermolysis bullosa
| Erythema multiforme | Erythema nodosum | Erythrasma
| Erythroderma
and exfoliative dermatitis | Eye bags
ECTHYMA
Ecthyma is a deep ulcerative
form of impetigo.
It can affect people of all ages but is more common in children,
elderly people and people with diabetes or who are immunocompromised
from disease or medication.
Cause
- Deep ulcerative form of impetigo caused by the streptococcal
and/or staphylococcal bacteria.
- Poor hygiene and malnutrition
may play a role.
Risk factors
- People with diabetes or who
are immunocompromised from disease or medication.
- Infection may follow an insect
bite or some minor skin trauma, including scratching.
- High temperature and humidity,
eg tropical places.
- Impetigo
- Symptoms
- A pustule (pushead) or blister
that rapidly enlarges, ulcerates and then developes a thick adherent
crust.
- Removal of the crust, which
can be difficult, leaves a deep punched out ulcer with a red
border.
- There may be associated lymphangitis
(red lines spreading upwards) and swollen lymph glands draining
the area.
- It usually affects the buttocks
and lower limbs.
- Pain.
- Heal within 6 - 8 weeks, often
leaving scars.
- Ecthyma gangrenosum is a less
common and serious form of ecthyma most commonly caused by the
pseudomonas bacteria. It usually occurs in patients who are critically
ill and immunocompromised. The characteristic lesion of ecthyma
gangrenosum a haemorrhagic (bloody) pustule that rapidly evolve
into a black gangrenous ulcer with a black/gray scab surrounded
by a red halo in as little as 12 hours. Ecthyma gangrenosum may
appear at any site but mainly affects the anogenital area and
armpits. The risk of septicaemia is very high so immediate hospitalisation
is necessary.
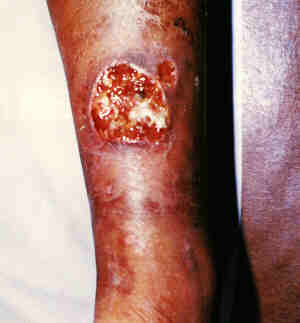 |
Ecthyma.
Click on
image for larger view |
- Complications
- Depressed oval or coin-shaped
scars often remain after ecthyma heals.
- In children. glomerulonephritis
(kidney inflammation) can result from streptococcal infection.
- Blood borne spread (septicaemia)
may develop in patuients who are immunosupprressed.
Diagnosis
- Skin swab for bacterial culture and sensitivity tests.
What you can do
- You should consult a doctor.
- Apply warm compresses
followed by removal of crusts.
- Clean with an antiseptic lotion
and apply a thin layer of the topical antibiotic prescribed by
the doctor.
- Observe careful personal hygiene.
- Do not share personal items
such as towels, shaving brushes and blades.
- Separate bed linens, towels,
etc., and boil separately.
- Advice should be given to
patients to avoid scratching itchy lesions. Keep fingernails short especially if you have
an itchy skin condtition such as eczema.
- Avoiding insects through use
of nets and repellent sprays.
What the doctor may do
- Prescribe oral and topical
antibiotics.
- Take a culture to help chose
the best antibiotic to use.
TOP
ECZEMA OR DERMATITIS
Eczema or dermatitis refers
to an inflammation of the skin characterised by redness, swelling,
weeping and scaling. It is usually itchy and constant scratching
leads to lichenification (leathery thickening of the skin). Doctors
divide eczemas into two broad groups:
Exogenous (exo means
external and gen means production in Greek) eczemas are
caused by external factors such as allergy to cement and plaster
or irritation from chemicals, soaps and detergents (see contact
dermatitis). Endogenous (endo means internal) eczemas,
on the other hand has to do with the skin's make-up or constitution.
Hence, they are also called constitutional eczemas. Endogenous
or constitutional eczemas cannot be cured whereas exogenous eczemas
can be if the causal substance can be avoided.
Symptoms
- Acute
eczema (or dermatitis) - red papules, red swollen, blistered,
crusted or excoriated skin.
- Chronic eczema (or dermatitis)
refers to a longstanding irritable area. It is dry, scaly, lichenified
(thick leathery often darkened) skin from chronic scratching.
- An in-between state is known
as subacute eczema.
- Itching is common to all stages
of eczema.
|
Stages |
Characteristics |
|
Acute |
Blisters, weeping, papules (pimply
bumps), pustules (pusheads) |
|
Subacute |
Redness, scaling, glistening serum
and crusting. |
|
Chronic |
Dryness, redness, scaling, lichenification
(leathery thickening of the skin with accentuation of the skin
markings) and fissuring (cracking) |
 |
Acute eczema.
Click on
image for larger view |
- Complications
- Complications
- Spread of eczema to other
areas of skin (autoeczematisation) or the entire body (see erythroderma).
- Secondary bacterial infection.
-
- Treatment
- See under specific types of
eczema.
TOP
Ectodermal dysplasia
(ED)
Ectodermal dysplasia
(ED) refers to a group of genetic disorders characterised by defects
in the hair, nails, teeth, skin, nails and sweat gland function.
Other tissues of ectodermal origin, such as the ears, eyes, lips,
mucous membranes of the mouth, nose, throat and central nervous
system, may be affected as well. The ectoderm is the outermost
layer of cells that forms the tissues mentioned above during feotal
life. All ectodermal dysplasias are present from birth and are
non-progressive. Dysplasia literally means “abnormal tissue
growth.”
Cause
Most cases are inherited although less common, EB occurs
without a family history, in which case it is due to spontaneous
mutation.
ED is caused by mutation or deletion of certain genes.
The different types of ectodermal
dysplasia are caused by the mutation or deletion of certain genes
located on different chromosomes. Because ectodermal dysplasias
are caused by a genetic defect they may be inherited or passed
on down the family line. In some cases, they can occur in people
without a family history of the condition, in which case a spontaneous
mutation has occurred.
The combination of physical
features a person has and the way in which it is inherited determines
if it is an ectodermal dysplasia. For example, hypohidrotic ectodermal
dysplasia affects the hair, teeth and sweat glands while Clouston
syndrome affects the hair and nails.
More than 180 different types
of ectodermal dysplasias exist. Yet, most types share some common
symptoms, ranging from mild to severe. The early diagnosis of
a specific type will help identify which combination of symptoms
the person has or will have.
Types
More than 180 subgroups have been described based on the presence
or absence of the four primary ectodermal dysplasia (ED) defects:The
180+ ectodermal dysplasias are recognized and named based on the
specific combination of symptoms shown in affected individuals.
The pattern of these features is important when a physician tries
to make a formal diagnosis.
The following are the most common types of ectodermal dysplasia
but it has to be said theat the largest group is diagnosed as
just Ectodermal Dysplasia, type unspecified.
Hypohidrotic (anhidrotic) ED
(HED) - inherited in one of three patterns: X-linked recessive,
autosomal recessive and autosomal dominant.
Ectrodactyly-Ectodermal Dysplasia-Clefting Syndrome (EEC) - Mutation
in the TP63 gene on chromosome 3q28. Autosomal dominant
Clouston syndrome - Also called hidrotic ectodermal dysplasia
because affected individuals sweat normally and exhibit no heat
intolerance. Autosomal dominant disorder.
Ankyloblepharon (fused eyelids) Ectodermal Dysplasia Clefting
(AEC Syndrome) - also known as Rapp-Hodgkin Syndrome. Hay-Wells
Syndrome is probably the same. Mutation in the TP63 gene on chromosome
3q28. Autosomal dominant.
Tooth nail syndrome
Focal Dermal Hypoplasia (FDH) (also known as Goltz syndrome)
Incontinentia Pigmenti (IP)
Symptoms
The signs and symptoms of ectodermal dysplasia depend on the structures
that are affected. These are often not apparent at birth and are
often only picked up during infancy or childhood.
Affected organ
Hair
Scalp and body hair may be thin, sparse, and light in colour
Hair may be coarse, excessively brittle, curly or even twisted
Nails
Fingernails and toenails may be thick, abnormally shaped, discoloured,
ridged, slow growing, or brittle
Sometimes nails may be absent
Cuticles may be prone to infection
Teeth
Abnormal tooth development resulting in missing teeth or growth
of teeth that are peg-shaped or pointed
Tooth enamel is also defective
Dental treatment is necessary and children as young as 2 years
may need dentures
Sweat glands
Eccrine sweat glands may be absent or sparse so that sweat glands
function abnormally or not at all
Without normal sweat production, the body cannot regulate temperature
properly
Children may experience recurrent high fever that may lead to
seizures and neurological problems
Overheating is a common problem, particularly in warmer climates
Other signs and symptoms include:
Lightly pigmented skin, in some cases red or brown pigment may
be present. Skin can be thick over the palms and soles and is
prone to cracking, bleeding and infection.
Skin may be dry and is prone to rashes and infection.
Dry eyes occur due to lack of tears. Cataracts and visual defects
may also occur.
Abnormal ear development may cause hearing problems.
Cleft palate/lip.
Missing fingers or toes (digits).
Respiratory infections due to lack of normal protective secretions
of the mouth and nose.
Foul smelling nasal discharge from chronic nasal infections.
Lack of breast development.
Treatment
There is no specific treatment
for ectodermal dysplasia. Management of the condition is by treating
the various symptoms. Patients often need to be treated by a team
of doctors and dentists, rather than a sole practitioner.
Patients with abnormal or no
sweat gland function should live in cooler climates or in places
with air conditioning at home, school and work. Cooling water
baths or sprays may be useful in maintaining a normal body temperature.
Artificial tears can be used to prevent damage to the cornea in
patients with defective tear production. Saline sprays can also
be helpful.
Saline irrigation of the nasal mucosa may help to remove purulent
debris and prevent infection.
Early dental evaluation and intervention is essential.
Surgical procedures such as repairing a cleft palate may lessen
facial deformities and improve speech.
Wigs may be worn to improve the appearance of patients with little
or no hair.
Most people with ectodermal
dysplasia can lead a full and productive life once they understand
how to manage their condition. Special attention must be paid
to children if sweating and mucous production abnormalities are
present. Recurrent high fevers may lead to seizures and neurological
problems.
Ehlers-Danlos syndrome
(EDS)
Ehlers-Danlos syndrome (EDS)
is the name given to a group of inherited disorders caused by
a defect in connective tissue synthesis and structure. This leads
to fragile, sagging skin, and loose joints. fragile tissue and
blood vessels. EDS may occur in males and females of all races
and usually first appears in young adults.
The Ehlers-Danlos syndromes
(EDS) are currently classified in a system of thirteen subtypes.
Each EDS subtype has a set of clinical criteria that help guide
diagnosis; a patient’s physical signs and symptoms will be
matched up to the major and minor criteria to identify the subtype
that is the most complete fit. There is substantial symptom overlap
between the EDS subtypes and the other connective tissue disorders
including hypermobility spectrum disorders, as well as a lot of
variability, so a definitive diagnosis for all the EDS subtypes—except
for hypermobile EDS (hEDS)—also calls for confirmation by
testing to identify the responsible variant for the gene affected
in each subtype.
Classical EDS (cEDS) - A final
diagnosis requires confirmation by molecular testing. More than
90% of those with cEDS have a heterozygous mutation in one of
the genes encoding type V collagen (COL5A1 and COL5A2). Rarely,
specific mutations in the genes encoding type I collagen can be
associated with the characteristics of cEDS. Classical EDS is
inherited in the autosomal dominant pattern.
Skin hyperextensibility and atrophic scarring; and
Generalized joint hypermobility (GJH).
Vascular EDS (vEDS) - Patients
with vEDS typically have a heterozygous mutation in the COL3A1
gene encoding type III collagen. Autosomal dominant.
Family history of vEDS with documented causative variant in COL3A1;
Arterial rupture at a young age;
Spontaneous sigmoid colon perforation in the absence of known
diverticular disease or other bowel pathology;
Uterine rupture during the third trimester in the absence of previous
C-section and/or severe peripartum perineum tears; and
Carotid-cavernous sinus fistula (CCSF) formation in the absence
of trauma.
A genetic defect causes reduced
amounts of collagen, disorganisation of collagen that is usually
organised into bundles, and alterations in the size and shape
of collagen. The type of EDS a patient has depends on how collagen
metabolism has been affected. For example vascular EDS is caused
by decreased or absent synthesis of type III collagen.
Skin hyperextensibility: it
is easy to pull the skin away from the body and once released
it retracts to its original state.
Skin fragility: the skin easily splits. Wounds heal very slowly
resulting in gaping fish-mouth or cigarette paper scars.
Epicanthic folds: skin folds between the eyes make the bridge
of the nose appear wide.
Molluscoid pseudotumours: small spongy lumps 2-3cm in diameter
over pressure points such as the knees and elbows.
Nodules: small, firm lumps just below the skin surface on the
arms and shins.
Hypermobility: joints bend more than usual. The fingers are most
often affected but all joints can be involved. The joints may
dislocate but can be popped back painlessly.
Bruising and haematomas may arise after trivial injuries.
Internal collagen defects: heart murmur (mitral valve prolapse)
and arterial/intestinal/uterine fragility or rupture (usually
associated with the Vascular Type).
scoliosis at birth and scleral fragility (associated with the
Kyphoscoliosis Type)
poor muscle tone (associated with the Arthrochalasia Type)
ELASTOSIS PERFORANS SERPIGINOSA (EPS)
This is a rare disorder in which
abnormal elastic tissue is being pushed out of the skin (a process
known as transepidermal elimination). It usually appears in early
childhood or young adulthood and males are more commonly affected.
Cause
- Part and parcel of other disorders
such as Ehler-Danlos syndrome, Marfan's syndrome, osteogenesis
imperfecta, pseudoxanthoma elasticum and Down syndrome.
- Drug induced, eg., due to
penicillamine, a drug used in the treatment of Wilson's disease
( a disorder of copper metabolism) and scleroderma.
Penicillamine interferes with the normal cross linking of elastic
tissue. The elastic tissue is therefore abnormal and the skin
tries to push it out of the skin.
- It is generally believed that
the abnormal elastic tissue in the dermis (inner skin) causes
irritation so the skin attempts to remove it through the epidermis
(outer skin).
- In drug-induced EPS due to
D-penicillamine (a drug used in the treatment of Wilson's disease),
abnormal elastic tissue is probably the result of penicillamine
inhibiting an enzyme responsible for the crosslinks within elastin.
However, in the case of non-drug induced EPS, the cause of abnormal
elastic tissue is unknown.
Symptoms
- Small red papules, grouped
in linear, annular (ring-like) or serpiginous (serpent-like)
patterns. The centre of these papules may contain a pit, which
is covered by a scaly plug.
- Usually there are no symptoms
but sometimes EPS can be itchy.
- Usually appears on the sides
of the neck, face and arms.
- Approximately 25 to 40 percent
of cases of EPS occur in association with genetic diseases such
as Ehler-Danlos syndrome, Marfan's syndrome, osteogenesis imperfecta,
pseudoxanthoma elasticum and Down syndrome.
Outlook
- EPS can persist for several
years before spontaneously resolving but has a tendency to recur.
What you can do
- You should consult a doctor.
What the doctor may do
- Determine the cause and eliminate
it (e.g. penicillamine)
- Treatment is not necessary
but the following may be used - topical corticosteroids, topical
retinoids, cryosurgery, and electrodesiccation and curettage.
TOP
Elastomas or elastin naevi include:
Isolated elastomas
Buschke-Ollendorf syndrome
Elastosis perforans serpiginosa
Buschke-Ollendorf syndrome is a rare hereditary disorder where
there is an increased accumulation of elastin in the dermis (elastoma).
Lesions may be present at birth but more usually appear within
the first year of life. They are firm yellowish wrinkled nodules
often group together to form plaques. The abdomen, back, buttock,
thighs or arms are commonly affected. Other manifestations of
the syndrome appear with time and may include osteopoikilosis
(inherited bone disorder identified on X-Ray), eye disorders and
spinal problems. autosomal dominant connective tissue disorder
due to mutations in the LEMD3 gene (607844) on chromosome 12q14.
Elastosis perforans serpiginosa
(EPS) is a perforating disorder. In this case, abnormal elastic
fibres are extruded through the epidermis. It presents in adolescence
and typically affects one or more sites on the face, neck and/or
arms. Groups of scaly papules are generally arranged in an arc
or ring shape. EPS is associated with other disorders of connective
tissue such as Marfan syndrome, Ehler Danlos syndrome, osteogenesis
imperfecta, pseudoxanthoma elasticum and Down syndrome.
Elephantiasis nostras
verrucosa (ENV)
Elephantiasis nostras verrucosa
(ENV) is a rare form of chronic lymphedema that causes progressive
cutaneous hypertrophy. It can lead to severe disfiguration of
body parts with gravity-dependent blood flow, especially the lower
extremities. Various factors can cause obstruction of the lymphatic
system and result in ENV. Clinically, ENV is characterized by
nonpitting edema and superimposed hyperkeratotic papulonodules
with a verrucose or cobblestone-like appearance. (Early-stage
lesions might exhibit pitting edema; late-stage lesions exhibit
nonpitting edema.) It needs to be differentiated from pretibial
myxedema, filariasis, lipedema, chromoblastomycosis, lipodermatosclerosis,
and venous stasis dermatitis (Table 1).1
edema and fibrosis of the skin.
Several conditions that block lymphatic drainage can induce lymphedema,
including neoplasms, trauma, radiation treatment, congestive heart
failure,1 obesity, hypothyroidism,6 chronic venous stasis,2 and
filarial infection.
The pathogenesis of ENV is still
unclear. It is conceivable that first the lymphatic channels are
damaged and blocked due to one or more of the above-mentioned
conditions, and excessive protein-rich fluid accumulates in the
dermis and subcutaneous tissues. Second, the protein-rich fluid
decreases oxygen tension and might decrease the immunity of the
skin. Third, poor immunity increases the skin’s susceptibility
to infection by micro-organisms. Finally, there is swelling, fibrosis,
and disfiguration of the affected areas.4 Hence, a vicious cycle
begins, as the underlying conditions predispose the skin to microbial
infections.
Elephantiasis nostras verrucosa
is commonly observed in gravity-dependent parts of the body, especially
in the lower extremities. In addition, other sites including the
upper extremities, abdomen, buttocks, face, or scrotum might be
involved.7–9 Elephantiasis nostras verrucosa usually begins
at the dorsal aspect of the foot and then progresses to the proximal
parts of the limbs. In the beginning, the lesion presents as mild
and persistent pitting edema. Later, the affected area loses its
elasticity and eventually has a hypertrophic, verrucose, cobblestone-like
appearance. During the physical examination, observation of the
Kaposi-Stemmer sign—inability to pinch the dorsal aspect
of skin at the base of the second toe5—is characteristic
of lymphedema. This phenomenon is attributed to skin thickening
caused by lymphedema.
Although the clinical presentation
is distinct, other diseases such as venous stasis dermatitis,
filariasis, lipedema, chromoblastomycosis, lipodermatosclerosis,
and pretibial myxedema should be clearly differentiated from ENV
(Table 1).1–5
The diagnosis of ENV is mainly
based on patient history, physical examination, and typical cutaneous
lesions. To identify causes of secondary lymphedema, skin biopsy
and imaging techniques including computed tomography, magnetic
resonance imaging, lymphangiography, and lymphoscintigraphy can
be necessary.
In the management of ENV, it
is crucial to treat the underlying causes. Lymphostasis can be
managed conservatively using medical bandages, compression stockings,
and mechanical massages. Elastic bandage compression is reported
to be an effective treatment.10 Diuretics and systemic antibiotics
might be needed to reduce edema and control infection. In addition,
hyperkeratotic plaques can be treated with topical keratolytics
or systemic retinoids.1 Owing to the teratogenicity of systemic
retinoids, it is important to provide contraception to female
patients before treatment. Patients should also receive careful
monitoring of serum lipids and liver function. Surgical intervention
can be considered in recalcitrant cases when the response to medical
treatment is poor.11 However, unsatisfactory outcomes are common
in the management of advanced stages of ENV.
Enlarged pores
Enlarged pores are depressions
in the facial skin surface that contain one or more openings to
the ducts carrying sweat and oil from their respective eccrine
glands and sebaceous glands.
Enlarged pores can be seen at
all ages and in all ethnic groups. Certain ethnic groups may have
larger pores, particularly those of African and Indian ancestry.
Pores often appear larger with age.
Factors that may lead to enlarged
pores include:
Increased sebum production
Hair follicle size
Use of comedogenic products
Loss of skin elasticity with age
Sun damage.
Acne is associated with enlarged pores — when sometimes open
comedones (blackheads) can be seen within a pore. Inflammatory
acne may cause enlarged pores through weakening sebaceous gland
and hair follicle openings, making them more prone to blockage.
Treatments that focus on preventing
and shrinking large pores are not very effective. They include:
Avoidance of skin creams that
induce blackheads and whiteheads
Weight loss (which is reported to reduce sebum production)
Chemical peels
Topical nicotinamide
Plant-derived copper chlorophyllin complexes
L-carnitine
Topical retinoids.
Oral treatments that are used for acne may also help. These include:
Combined oral contraceptives
Spironolactone
Isotretinoin.
Physical treatments targeting
the sebaceous glands may help enlarged pores. These include:
Laser treatment
Radiofrequency microneedling (skin needling).
Eosinophilic cellulitis
(Wells syndrome)
What is Wells syndrome?
Wells syndrome is a rare condition
of unknown cause. It is also called ‘eosinophilic cellulitis’.
What does Wells syndrome look
like?
Typically the rash is preceded
by itching or burning skin and consists of markedly swollen nodules
and plaques (lumps) with prominent borders. The patches are usually
bright red at first, frequently looking like cellulitis, then
fade over four to eight weeks, leaving green, grey or brown patches.
They can blister. The rash most commonly occurs on the limbs,
but may also affect the trunk.
The patient often feels very
tired and has a fever in approximately 25% of cases.
Wells syndrome
Wells syndrome
Investigations
A blood count may reveal increased
numbers of white blood cells called eosinophils – these are
often associated with allergy or insect bites.
The diagnosis of Wells syndrome
can be established by a skin biopsy finding of typical histopathological
features with many eosinophils and characteristic ‘flame
figures’. However, flame figures are not diagnostic of Wells
syndrome and can be seen in other conditions that have increased
numbers of eosinophils.
An important part of the management
of patients with Wells syndrome is to exclude underlying causes
such as parasitic disoders (e.g. a worm infestation) or an allergic
contact dermatitis with the help of the appropriate tests.
Treatment
Oral corticosteroid treatment
with prednisone can lead to a dramatic improvement within days
and the course is typically tapered over one month. Other treatments
include minocycline, dapsone, griseofulvin, ciclosporin and oral
antihistamines.
Mild cases may respond to topical
steroid therapy alone.
eosinophilic fasciitis
Eosinophilic fasciitis is a
rare scleroderma-like disorder characterised by inflammation,
swelling and thickening of the skin and fascia (fibrous tissue
that separates different layers of tissues under the skin). It
affects the forearms, the upper arms, the lower legs, the thighs,
and the trunk (in order of decreasing frequency). The disease
is considered by some to be a deep variant of the skin condition,
morphoea.
Eosinophilic fasciitis
Eosinophilic fasciitis
What causes eosinophilic fasciitis and who gets it?
The cause of eosinophilic fasciitis
is unknown but it may have something to do with abnormal immune
responses as hypergammaglobulinaemia and antinuclear antibodies
are present. In addition, toxic, environmental, or drug exposures
have been implicated.
It affects females and males,
children and adults, with most cases occurring between the ages
of 30 and 60 years.
What are the clinical features
of eosinophilic fasciitis?
Patients usually present suddenly
with painful, tender, swollen and red extremities. Within weeks
to months patients develop stiffness and affected skin becomes
indurated, creating a characteristic orange-peel appearance over
the surfaces of the extremities. In severely affected areas, the
skin and the subcutaneous tissue are bound tightly to the underlying
muscle. This creates a woody-type appearance.
In 50% of cases, the disease
is precipitated by an episode of strenuous physical exercise or
activity. Other signs and symptoms that may be present include:
Malaise, weakness, fever and
weight loss
Joint contractures of the elbows, wrists, ankles, knees and shoulders
Carpal tunnel syndrome
Inflammatory arthritis
How is eosinophilic fasciitis diagnosed?
Laboratory studies in early
active disease show eosinophilia, elevated ESR and polyclonal
immunoglobulin G. However, full thickness skin biopsy that includes
the dermis, subcutaneous fat and fascia is necessary to confirm
a diagnosis of eosinophilic fasciitis, which has specific pathological
features.
What is the treatment for eosinophilic
fasciitis?
Treatment of eosinophilic fasciitis
is directed at preventing tissue inflammation. Oral corticosteroids
are the mainstay of treatment, with most patients responding well
to moderate-to-high doses of corticosteroids particularly if started
early in the course of the disease. Continued low doses may be
required for 2-5 years. In some cases, the disease may resolve
spontaneously.
Other drugs that may be used
in conjunction with corticosteroids include:
Nonsteroidal anti-inflammatory
agents (NSAIDs)
Hydroxychloroquine
Colchicine
Cimetidine
Azathioprine
Cyclosporine
Cyclophosphamide
Methotrexate
A physiotherapist is also important in the overall management
of eosinophilic fasciitis to prevent and treat joint contractures.
Eosinophilic folliculitis
Eosinophilic folliculitis is
a recurrent skin disorder of unknown cause. It is also known as
"eosinophilic pustular folliculitis" or "Ofuji
disease". Skin biopsies of this disorder find eosinophils
(a type of immune cell) around hair follicles – hence its
name.
There are several variants of
eosinophilic folliculitis.
All of them present with itchy
papules (bumps) or pustules. Eosinophilic folliculitis is rare
and more often affects males than females. Variants include:
Classic type – this occurs
most commonly in Japan
Eosinophilic folliculitis associated with advanced Human Immunodeficiency
Virus (HIV) infection
Infantile eosinophilic folliculitis
Cancer-associated eosinophilic folliculitis
Medication-associated eosinophilic folliculitis
What does eosinophilic folliculitis look like?
Eosinophilic folliculitis presents
with red or skin-coloured dome shaped papules (bumps) and pustules.
It may look rather like acne or other forms of folliculitis. The
papules mostly appear on the face, scalp, neck and trunk and may
persist for weeks or months. Less commonly, urticarial lesions
are seen (these are larger red irritable wheal-like patches similar
to urticaria). Palms and soles may rarely develop similar papules
and pustules, but in such cases the condition should not be called
"folliculitis" as there are no follicles in these areas.
Longstanding cases may develop
dermatitis or a form of prurigo, presumably because of the itching
and scratching.
Eosinophilic folliculitis of
HIV
Eosinophilic folliculitisEosinophilic folliculitisEosinophilic
folliculitisEosinophilic folliculitis
How is eosinophilic folliculitis diagnosed?
Skin biopsy reveals eosinophils
under the skin surface and around the hair follicles and sebaceous
glands. In many cases blood tests show a mild rise in eosinophil
cells and immunoglobulin-E (IgE), and reduced IgG and IgA levels.
Eosinophilic folliculitis is
often a feature of immunodeficiency. Eosinophilic folliculitis
associated with HIV infection presents when levels of CD4 lymphocyte
cells drop below 300 cells/mm3, a level at which there is an increased
risk of a secondary opportunistic infection. Cases of eosinophilic
folliculitis have also reported after bone marrow transplantation
before the immune system is back to normal functioning, and in
some individuals with inherited immune deficiencies.
What is the cause of eosinophilic
folliculitis of HIV?
The cause of eosinophilic folliculitis
of HIV is not known. Immunodeficiency appears to lead to increased
risk of allergic-type skin diseases. There is no proof that bacterial,
fungal or viral secondary infection is the cause, although some
researchers have postulated overgrowth of malassezia or demodex
(the hair follicle mite) might be involved. Another theory is
that there is a change in the immune system causing eosinophils
to attack the sebum (oils produced in the skin) of sebaceous gland
cells.
What is the treatment for eosinophilic
folliculitis?
In patients with HIV, eosinophilic
folliculitis is likely to improve or resolve with HAART (Highly
Active Anti-Retroviral Treatment), as CD4 cell counts rise above
250/mm3.
Other treatments that may be
effective include:
Indomethacin and other nonsteroidal
anti-inflammatory drugs are reported effective in up to 70% of
cases of eosinophilic folliculitis
Dapsone
Tetracycline antibiotics
Other antibiotics including metronidazole
Phototherapy
Topical steroids
Calcineurin inhibitors such as tacrolimus ointment
Oral antihistamines such as cetirizine
Colchicine
Itraconazole
Permethrin cream (topical insecticide)
Nicotine patches
Isotretinoin and acitretin.
EPHELIDES
Ephelides are freckles most
commonly seen in children with fair skins, especially those of
Celtic origin.
Cause
- Inherited tendency. It's been
found that caucasians with many ephilides have at least one copy
of a variant MC1R (melanocortin 1 receptor) gene, which is the
same variant that causes red hair. The MC1R gene is responsible
for producing a set of instructions to MC1R protein, which sits
on the surface of the melanocytes (melanin pigment producing
cells). When working properly, it turns pheomelanin into eumelanin,
the pigment responsible for hair colors other than red. In people
with numerous ephilides and red hair, the variant MC1R gene does
not work properly so melanin production switches to generating
phaemelanin by default. A different variant may be responsible
for ephilides in non-caucasians.
Symptoms
- Small light brown or tan spots
on the sun-exposed skin, especially the cheeks, nose, shoulders
and the upper back.
- They are not present at birth
and appear later in childhood, with sun-exposure.
- Ephilides are more common
in people with fair skin of any race. They occur in especially
large numbers in people with white skin that cannot tan (Fitzpatrick
skin phototype 1) and red hairs (Celtic ancestry).
- Freckles typically become
darker with sun exposure for example, during the summer and lighten
during the winter months.
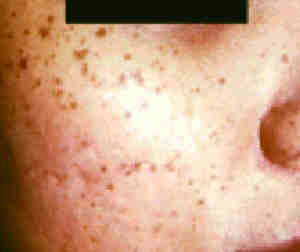 |
Ephelides.
Click on
image for larger view |
What you can do
- Protect against the sun (see
sun protection).
- Consult a doctor for treatment
if the freckles bother you.
What the doctor may do
- Lighten with chemical
peels or liquid nitrogen aplications.
- Prescribe hydroquinone
containing lightening creams.
TOP
Epidermoid cyst
A cyst is a benign, round, dome-shaped
encapsulated lesion that contains fluid or semi-fluid material.
It may be firm or fluctuant and often distends the overlying skin.
There are several types of cyst. The most common are described
here.
What is a pseudocyst?
Cysts that are not surrounded
by a capsule are better known as pseudocysts. These commonly arise
in acne.
Who gets cysts?
Cysts are very common, affecting
at least 20% of adults. They may be present at birth or appear
later in life. They arise in all races. Most types of cyst are
more common in males than in females.
What causes cysts?
The cause of many cysts is unknown.
Epidermoid cysts are due to
proliferation of epidermal cells within dermis. Their origin is
the follicular infundibulum. Multiple epidermoid cysts may indicate
Gardner syndrome.
An epidermal inclusion cyst is a response to an injury. Skin is
tucked in to form a sac that is lined by normal epidermal cells
that continue to multiply, mature and form keratin.
The origin of a trichilemmal cyst is hair root sheath. Inheritance
is autosomal dominant (the affected gene is within short arm of
chromosome 3) or sporadic.
The origin of steatocystoma is the sebaceous duct within the hair
follicle. Steatocystoma multiplex is sometimes an autosomal dominantly
inherited disorder due to mutations localised to the keratin 17
(K17) gene, when it may be associated with pachyonychia congenita.
More often, steatocysts are sporadic, when these mutations are
not present.
The origin of the eruptive vellus hair cyst is follicular infundibulum.
It may be inherited an autosomal dominant disorder due to mutations
in keratin gene.
A dermoid cyst is a hamartoma, a developmental error.
The origin of a ganglion cyst is degeneration of the mucoid connective
tissue of a joint.
Occlusion of pilosebaceous units (hair follicles) or eccrine sweat
ducts leads to a build-up of secretions. This can present as milia.
Occlusion of the orifice of a mucous gland can lead to a fluid-filled
cyst in a mucous membrane (lip, vulva, vagina).
A milium is a pseudocyst due to failure to release keratin from
an adnexal structure. The origin of primary milium is infundibulum
of vellus hair follicle at the level of the sebaceous gland; a
tiny version of an epidermoid cyst. The origin of secondary milium
is a retention cyst within a vellus hair follicle, sebaceous duct,
sweat duct or epidermis.
Pseudocysts in acne are formed by occlusion of the follicle by
keratin and sebum.
What are the clinical features of cysts?
Epidermoid cyst
Epidermoid cysts occur on face,
neck, trunk or anywhere where there is little hair.
Most epidermoid cysts arise in adult life.
They are more than twice as common in men as in women.
They present as one or more flesh–coloured to yellowish,
adherent, firm, round nodules of variable size.
A central pore or punctum may be present.
Keratinous contents are soft, cheese-like and malodorous.
Scrotal and labial cysts are frequently multiple and may calcify.
Epidermoid cyst is also called follicular infundibular cyst, epidermal
cyst, keratin cyst.
Epidermoid cyst
CystCystGardner syndrome
Gardner syndrome
More images of epidermoid cysts ...
Trichilemmal cyst
90% of trichilemmal cysts occur
on scalp; otherwise face, neck, trunk, and extremities.
Most trichilemmal cysts arise in middle age.
In 70% of cases, trichilemmal cysts are multiple.
They presents as adherent, round or oval, firm nodules.
There is no punctum.
The keratinous content is firm, white and easily enucleated.
A trichilemmal cyst is also called pilar cyst.
Trichilemmal cyst
Pilar cystPilar cystsPilar cyst
More images of epidermoid cysts ...
Steatocystoma
A solitary steatocystoma is
known as steatocystoma simplex.
More often, there are multiple lesions (steatocystoma multiplex)
on chest, upper arms, axillae and neck.
The cysts arise in the late teens and 20s due to the effect of
androgens, and persist lifelong.
They are freely moveable, smooth flesh to yellow colour papules
3–30 mm in diameter.
There is no central punctum.
Content of cyst is predominantly sebum.
Steatocystoma multiplex
SteatocystSteatocystoma multiplexSteatocystoma multiplex
Eruptive vellus hair cysts
Eruptive vellus hair cysts are
present in childhood if familial, and later if sporadic.
Multiple 2–3 mm papules develop over the sternum.
The cysts contain vellus hairs.
Dermoid cyst
A cutaneous dermoid cyst may
include skin, skin structures and sometimes teeth, cartilage and
bone.
Most dermoid cysts are found on face, neck, scalp; often around
eyelid, forehead and brow.
It is a thin-walled tumour that ranges from soft to hard in consistency.
The cyst is formed at birth but the patient may not present until
an adult.
Dermoid cysts
Dermoid cystDermoid cystDermoid cyst
Ganglion cyst
A ganglion cyst most often involves
scapholunate joint of dorsal wrist.
These arise in young to middle-aged adults.
They are 3 times more common in women than in men.
The cyst is a unilocular of multilocular firm swelling 2–4
cm in diameter that transilluminates.
Cyst contents are mainly hyaluranic acid, a golden-coloured goo.
Ganglion cyst
Ganglion cystGanglion cystGanglion cyst
Mucous/myxoid pseudocysts arise in older adults on distal phalanx
They arise from distal interphalangeal joint, associated with
osteoarthritis.
They often present as a longitudinal depression in the nail due
to compression on the proximal matrix.
Myxoid pseudocyst
Digital myxoid cystMyxoid cystDigital myxoid cyst
More images of digital myxoid pseudocysts ...
Labial mucous/myxoid cyst
A cyst in the lip may be due
to occlusion of the salivary duct
They are also called mucocoele.
It is a soft to firm firm, 5–15 mm diameter, semi-translucent
nodule.
Mucocoele of lip
Mucocoele of the lipMucocoeleMucocoele
Hidrocystoma
Hidrocystoma is a translucent
jelly-like cyst arising on an eyelid.
It is also known as cystadenoma, Moll gland cyst, and sudoriferous
cyst.
The common solitary translucent eyelid cyst is an apocrine hidrocystoma.
Multiple cysts on the lower eyelid are eccrine hidrocystomas.
Hidrocystoma of eyelid
HidrocystomaHidrocystomaHidrocystoma
More images of hidrocystoma of eyelid ...
Milium/milia
Milia are 1–2 mm superficial
white dome-shaped papules containing keratin
Primary milia arise in neonates (50%), adolescents and adults;
they are rarely familial and sometimes eruptive.
Primary milia occur on eyelids, cheeks, nose, mucosa (Epstein
pearls) and palate (Bohn nodules) in babies; and eyelids, cheeks
and nose of older children and adults.
Transverse primary milia are sometimes noted across nasal groove
or around areola.
In milia en plaque, multiple milia arise on an erythematous plaque
on face, chin or ears.
Secondary milia arise at the site of epidermal repair after blistering
or injury, eg epidermolysis bullosa, bullous pemphigoid, porphyria
cutanea tarda, thermal burn, dermabrasion.
Secondary milia are reported as an adverse effect of topical steroids,
5-fluorouracil cream, vemurafenib and dovitinib.
Milia
MiliaEyelid miliaMilia
More images of milia ...
Vulval mucous cyst
A vulval mucous cyst is due
to occlusion of Bartholin or Skene duct.
It presents as a soft swelling in the introitus of vagina: a posterior
swelling is a Bartholin cyst and a periurethral swelling is a
Skene cyst.
Comedo and acne pseudocyst
Comedones are pseudocysts formed
by occlusion of follicle by keratin and sebum.
The open comedo (whitehead) and closed comedo (blackhead) are
small, superficial papules typical of acne vulgaris
Solar comedones arise in sun-damaged skin and are associated with
smoking.
Large uninflamed pseudocysts accompany inflammatory nodules in
nodulocystic acne and hidradenitis suppurativa.
Comedones
Open comedones
Open comedones
Closed comedones
Closed comedones
Solar comedones
Solar comedones
Pseudocyst of auricle
Pseudocyst of auricle (external
ear) follows trauma.
Pseudocyst of auricle
Auricular pseudocyst
Complications of cysts
Rupture of a cyst
The contents of the cyst may
penetrate the capsular wall and irritate surrounding skin.
The area of tender, firm inflammation spreads beyond the encapsulated
cyst.
Sterile pus may be discharged.
Secondary infection
A ruptured cyst may infrequently
become secondarily infected by Staphylococcus aureus, forming
a furuncle (boil).
Pressure effect
A dermoid cyst can cause pressure
on underlying bony tissue.
A ganglion cyst can cause joint instability, weakness, limitation
of motion and may compress a nerve.
A digital mucous cyst may place pressure on the proximal matrix
and cause malformation of the nail.
Malignancy
Cutaneous cysts and pseudocysts
are non-proliferative benign lesions.
Nodulocystic basal cell carcinoma is a common skin cancer that
presents as a rounded nodule and may initially be mistaken for
a cyst, but steady enlargement, destruction of the epidermis with
ulceration and bleeding occur eventually.
Malignant proliferative trichilemmal cyst is actually a misnomer.
It is an extremely rare tumour.
How are cysts diagnosed?
Cysts are usually diagnosed
clinically as they have typical characteristics. When a cyst is
surgically removed, it should undergo histological examination.
The type of lining of the wall of cyst and the cyst contents help
the pathologist classify it.
Epidermoid cysts are lined with
stratified squamous epithelium that contains a granular layer.
Laminated keratin contents are noted inside the cyst. An inflammatory
response may be present in cysts that have ruptured.
Trichilemmal cysts have a palisaded peripheral layer without granular
layer. Contents are eosinophilic hair keratin. Older cysts may
exhibit calcification. The proliferating variety is considered
a tumour.
Steatocystoma has a folded cyst wall with prominent sebaceous
gland lobules.
Dermoid cyst contains fully mature elements of the skin including
fat, hairs, sebaceous glands, eccrine glands, and in 20%, apocrine
glands.
The lining of the wall of a ganglion cyst or digital mucous cyst
is collagen and fibrocytes. It contains hyalin material.
Hidrocystoma has a thin lining wall of eosinophilic bilaminar
cells.
What is the treatment for cysts?
Asymptomatic epidermoid cysts
do not need to be treated. In most cases, attempt to remove only
the contents of a cyst is followed by recurrence. If desired,
cysts may be fully excised. Recurrence is not uncommon, and re-excision
may be surgically challenging.
Inflamed cysts are sometimes
treated with:
Incision and drainage
Intralesional injection with triamcinolone
Oral antibiotics
Delayed excision biopsy
How can cysts be prevented?
Unknown.
What is the outlook for cysts?
Cysts generally persist unless
surgically removed.
EPIDERMAL
NAEVI
Epidermal naevi are developmental
abnormalities caused by an overgrowth of the epidermis (upper
layers of the skin). They appear at birth or during childhood,
usually in the first year of life. The abnormality arises from
a defect in the ectoderm. This is the outer layer of the embryo
that gives rise to epidermis and neural tissue. Epidermal nevus
is a clinical term for a family of skin lesions that involve the
outer portion of skin, the epidermis, and are distributed in a
linear and often swirled pattern. Overall, epidermal nevi are
not uncommon congenital malformations, occurring in 1-3 per 1000
births.
Cause
- There are two copies of every
gene, one derived from the individual's mother and the other
from their father. There are two populations of skin cells (a
situation referred to as mosaicism), containing either the mother
or the father's genes. The epidermal naevus is thought to the
product of the abnormal skin cell If one of these populations
is abnormal, an epidermal naevus developed. Luckily, epidermal
naevi very rarely affect more than one member of the family.
Mutations have been detected in FGFR3, PIK3CA and HRAS in epidermal
naevi. Epidermal naevi are distributed along the lines of Blashko.
These lines are the tracks taken by groups of genetically identical
cells in the developing embryo. Skin cells that have the active
abnormal gene spread out to form the epidermal naevus, whereas
the remaining skin cells form the other areas of apparently normal
skin. New research has found point mutations in keratin genes
that support this theory. The abnormal gene is found in the epidermal
naevus cells but not in the normal skin. The same keratin 1 and
keratin 10 gene abnormalities have been found in parents who
have epidermolytic epidermal naevus and in their offspring who
have bullous ichthyosiform erythroderma (a rare form of ichthyosis).
So the epidermolytic epidermal naevus is thought to be a mosaic
form of this type of ichthyosis.The ATP2A2 gene abnormality that
arises in Darier disease has been detected in the affected cells
of a patient with acantholytic epidermal naevus, so this type
may be a mosaic form of Darier disease. Linear porokeratosis
may be a mosaic form of disseminated superficial actinic porokeratosis.
- Epidermal naevi are believed
to be genetically ‘mosaic’, meaning that the mutation
causing the nevi are not found in other cells of the body. Mosaicism
arises when the genetic mutation occurs in one of the cells of
the early embryo sometime after conception; such mutations are
called ‘somatic’ mutations. This mutated cell, like
the other normal cells, continues to divide and gives rise to
mutated daughter cells that will populate a part of the body.
The linear patterning of the epidermal nevus reflects the movement
of the mutant daughter cells during fetal growth. These linear,
developmental patterns are termed the ‘lines of Blaschko’.
Many epidermal cells within these affected areas harbor the mutant
gene, while most or all cells from uninvolved areas do not. After
birth, the nevus “grows with the child”, although some
new areas of involvement and/or extension of the nevus to new
areas can occur.
- Symptoms
- Raised brown area with a rough
warty surface.
- Tendency to occur in lines
along the length of a limb.
The lesions may be single or multiple and are usually present
at birth. All epidermal nevi show some changes in texture which
can range from very rough, warty and spiny, and often darker
than the surrounding normal or uninvolved skin (verrucous epidermal
nevus), to red and scaly (inflammatory linear verrucous epidermal
nevus or ILVEN), to yellowish, rough and pebbly appearance due
to proliferation of oil- or ’sebaceous’ gland-like
structures (nevus sebaceous). If they are limited to the epidermal
linings of the hair follicles, they may appear like blackheads:
nevus comedonicus. Another related and relatively common entity,
the Becker’s nevus, is a form of epidermal nevus with a
somewhat velvety texture, mild to moderate hyperpigmentation
and coarser vellus hairs. It typically occurs on the trunk during
childhood or adolescence in a broader, patchy rather than linear
pattern. The term ‘nevus’ is also used for other birthmarks,
malformations and some benign growths, such as melanocytic nevi,
or ‘moles’.
What you can do
- You should consult a doctor.
What the doctor may do
- Destroy the area using electrosurgery or the
carbon dioxide laser.
- Excise the area with or without
grafting.
TOP
EPIDERMOLYSIS BULLOSA (EB)
Epidermolysis bullosa (EB) is
a group of rare inherited disorders characterised by blistering
of the skin after minor trauma. Some forms of EB are mild with
few blisters but others may be more severe with many blisters
on the skin and even inside the body such as the mouth, oesophagus,
stomach, respiratory tract, bladder, and elsewhere.There several
types of epidermolysis bullosa.
Cause
Most cases of EB are inherited although less common, EB occurs
without a family history, in which case it is due to spontaneous
mutation. Inheritence may occur in 2 ways:
- Autosomal dominant where only
one parent need be affected and the offspring has a 50% chance
of inheriting the defect. In autosomal dominant EB, only one
abnormal gene is needed to express the disease.
- Autosomal recessive where
both parents have to be carriers and the offspring has a 25%
chance of inheriting the defect. In autosomal recessive EB, you
must have two EB genes (one from each parent) to have the disease.
If you only inherit one abnormal recessive gene, you will be
a carrier but will not express the disease.
Types of EB
There are many other subtypes of EB. The presentation and severity
of EB is affected by the specific genetic changes and can at
times be difficult to classify.
- Epidermolysis bullosa simplex
(EBS) of which there are 3 subtypes
- Localised EBS (Weber-Cockayne
type) - Blisters develop on hands and feet.
- Generalised EBS (Koebner type)
- Blisters appear all over the body but commonly on hands, feet
and extremities.
- Generalised severe EBS (Dowling
Meara type) - Blisters appear all over the body and may involve
the mouth, gastrointestinal and respiratory tract. May be fatal.
- Junctional epidermolysis bullosa
(JEB) of which there are 2 subtypes
- Generalised severe JEB (Herlitz
type) - Generalised and most severe form of JEB where
blisters appear all over the body and often involve mucous membranes
and internal organs. High risk of fatality, usually within the
first 1 - 2 years of life.
- Intermediate JEB (Non-Herlitz
type) - Generalised blistering and mucosal involvement present
at birth or soon after. Less risk of fatality and survivors improve
with age.
- Dystrophic epidermolysis bullosa
(DEB) which may be of 3 subtypes
- Dominant generalised DEB and
it's variant Barts syndrome which is associated with aplasia
cutis
- Generalised severe recessive
(R) DEB (Hallopeau-Siemens type)
- Generalised intermediate RDEB
(Non-Hallopeau-Siemens type)
- Kindler syndrome
Who gets EB
Symptoms
- In mild types blisters appear
on the hands and feet and elbows and knees.
- In more severe cases, they
appear all over the body and healing causes fusion of digits,
contraction of joints, narrowing of oesophagus.
- In very severe forms, the
blisters are associated with may occur anywhere on the body but
most commonly at sites exposed to friction and minor trauma such
as the feet and hands and knees and elbows.
- Usually appear at or soon
after birth but in mild cases, it may not be noticed until the
child is older. Sometimes, mild cases may be mistaken as ordinary
friction induced blisters.
- In severe cases, the baby
is born with raw skin because the blisters were shed in the womb.
Complications
- Infection.
- Scarring.
- Fusion of digits (fingers
and toes),
- Contractures of joints.
- Problems feeding due to narrowing
of oesophagus.
- Death
Diagnosis
- Based of family history and
clinical signs.
- Skin biopsy of a newly induced
blister which undergoes immunofluorescence antigen mapping (IFM)
and/or transmission electron microscopy (EM).
- Genetic testing may be available
in some countries.
- What you can do
- You should consult a doctor.
- Protect the skin against trauma.
- What the doctor may do
- There is no cure for EB.
- Treatment is symptomatic,
the main aims being to protect the skin against trauma, promote
healing and prevent complications.
- Give genetic counseling.
- Treat the complications.
- Research in gene therapy and
cell-based therapy is continuing in an effort to improve the
quality of life.
TOP
erysipelas
- what is erysipelas?
- Erysipelas is a superficial
form of cellulitis, a potentially serious bacterial infection
affecting the skin.
- Erysipelas affects the upper
dermis and extends into the superficial cutaneous lymphatics.
It is also known as St. Anthony's fire, with reference to the
intense rash associated with it.
- Who gets erysipelas?
- Erysipelas most often affects
infants and the elderly, but can affect any age group. Risk factors
are similar to those for other forms of cellulitis. They may
include:
- Previous episode(s) of erysipelas
Breaks in the skin barrier due insect bites, ulcers and chronic
skin conditions such as psoriasis, athlete’s foot and eczema
Current or prior injury (eg trauma, surgical wounds, radiotherapy)
In newborns, exposure of the umblical cord and vaccination site
injury
Nasopharyngeal infection
Venous disease (eg gravitational eczema, leg ulceration) and/or
lymphoedema
Immune deficiency or compromise, such as
Diabetes
Alcoholism
Obesity
Human immunodeficiency virus (HIV)
Nephrotic syndrome
Pregnancy
What causes erysipelas?
- Unlike cellulitis, almost
all erysipelas is caused by Group A beta haemolytic streptococci
(Streptococcus pyogenes). Staphylococcus aureus, including methicillin
resistant strains (MRSA),Streptococcus pneumoniae,Klebsiella
pneumoniae,Yersinia enterocolitica, andHaemophilus influenzae
have also been found to cause erysipelas.
- What are the clinical features
of erysipelas?
- Symptoms and signs of erysipelas
are usually abrupt in onset and often accompanied by fevers,
chills and shivering.
- Erysipelas predominantly affects
the skin of the lower limbs, but when it involves the face it
can have a characteristic butterfly distribution on the cheeks
and across the bridge of the nose.
- The affected skin has a very
sharp, raised border.
It is bright red, firm and swollen. It may be finely dimpled
(like an orange skin).
It may be blistered, and in severe cases may become necrotic.
Bleeding into the skin may cause purpura.
Cellulitis does not usually exhibit such marked swelling, but
shares other features with erysipelas, such as pain and increased
warmth of affected skin.
In infants, it often occurs in the umbilicus or diaper/napkin
region.
Bullous erysipelas can be due to co-infection with Staphylococcus
aureus (including MRSA).
Erysipelas
ErysipelasErysipelasErysipelasErysipelas
What are the complications of erysipelas?
- Erysipelas recurs in up to
one third of patients due to:
- Persistence of risk factors
Lymphatic damage (hence impaired drainage of toxins)
Complications are rare but can include:
- Abscess
Gangrene
Thrombophlebitis
Chronic leg swelling
Infections distant to the site of erysipelas:
Infective endocarditis (heart valves)
Septic arthritis
Bursitis
Tendonitis
Post-streptococcal glomerulonephritis (a kidney condition affecting
children)
Cavernous sinus thrombosis (dangerous blood clots that can spread
to the brain)
Streptococcal toxic shock syndrome (rare)
How is erysipelas diagnosed?
- Erysipelas is usually diagnosed
by the characteristic rash. There is often a history of a relevant
injury. Tests may reveal:
- Raised white cell count
Raised C-reactive protein
Positive blood culture identifying the organism
MRI and CT are undertaken in case of deep infection.
- What is the treatment for
erysipelas?
- General
- Cold packs and analgesics
to relieve local discomfort
Elevation of an infected limb to reduce local swelling
Compression stockings
Wound care with saline dressings that are frequently changed
Antibiotics
- Oral or intravenous penicillin
is the antibiotic of first choice.
Erythromycin, roxithromycin or pristinamycin may be used in patients
with penicillin allergy.
Vancomycin is used for facial erysipelas caused by MRSA
Treatment is usually for 10–14 days
What is the outlook for erysipelas?
- While signs of general illness
resolve within a day or two, the skin changes may take some weeks
to resolve completely. No scarring occurs.
- Long term preventive treatment
with penicillin is often required for recurrent attacks of erysipelas.
- Erysipelas recurs in up to
one third of patients due to persistence of risk factors and
also because erysipelas itself can cause lymphatic damage (hence
impaired drainage of toxins) in involved skin which predisposes
to further attacks.
- If patients have recurrent
attacks, long term preventive treatment with penicillin may be
considered.
-
- erythema ab igne
- What is erythema ab igne?
- Erythema ab igne (EAI) is
a skin reaction caused by chronic exposure to infrared radiation
in the form of heat. It was once a common condition seen in the
elderly who stood or sat closely to open fires or electric space
heaters. Although the introduction of central heating has reduced
EAI of this type, it is still found in individuals exposed to
heat from other sources.
- What are the signs and symptoms
and who is at risk?
- Limited exposure to heat,
insufficient to cause a direct burn, causes a mild and transient
red rash resembling lacework or a fishing net. Prolonged and
repeated exposure causes a marked redness and colouring of the
skin (hyper- or hypo-pigmentation). The skin and underlying tissue
may start to thin (atrophy) and rarely sores may develop. Some
patients may complain of mild itchiness and a burning sensation.
- Erythema ab igne
Erythema ab igneErythema ab igneErythema ab igneErythema ab igne
Localised lesions seen today reflect the different sources of
heat that people may be exposed to. Examples include:
- Repeated application of hot
water bottles or heat pads to treat chronic pain, e.g. chronic
backache
Repeated exposure to car heaters or furniture with built-in heaters
Occupational hazard for silversmiths and jewellers (face exposed
to heat), bakers and chefs (arms)
What treatments are available?
- The source of chronic heat
exposure must be avoided. If the area is only mildly affected
with slight redness, the condition will resolve by itself over
several months. If the condition is severe and the skin pigmented
and atrophic, resolution is unlikely. In this case, there is
a possibility that squamous cell carcinomas may form. If there
is a persistent sore that doesn't heal or a growing lump within
the rash, a skin biopsy should be performed to rule out the possibility
of skin cancer. Abnormally pigmented skin may persist for years.
Treatment with topical tretinoin or laser may improve the appearance.
- erythema dyschromicum
perstans
-
- What is erythema dyschromicum
perstans?
- Erythema dyschromicum perstans
is also called ashy dermatosis (of Ramirez), because of its colour.
It is a benign skin condition characterised by well-circumscribed
round to oval or irregular patches on the face, neck and trunk
that are grey in colour. They may be symmetrical in distribution
or unilateral.
- Early lesions may be reddish
in colour, often with a more pronounced border, and they may
be somewhat elevated. However, this phase is not always observed.
- The patient is otherwise well
with no associated disease or blood test abnormality.
- Erythema dyschromicum perstans
Erythema dyschromicum perstansErythema dyschromicum perstansErythema
dyschromicum perstans
Who gets erythema dyschromicum perstans?
- Erythema dyschromicum perstans
most often affects darker skinned patients, most frequently Latin
Americans and Indians. However it has also been reported in people
of lighter skin colour and various ethnicities.
- Erythema dyschromicum perstans
may occur at any age but it appears to be more frequent in young
adults. Women are affected more often than men.
- What causes erythema dyschromicum
perstans?
- The exact cause for the disease
remains unidentified. It is often classified as a variant of
lichen planus because of its histopathological features. Over
the years, theories have included:
- Genetic susceptibility
Toxic effects of chemicals such as ammonium nitrate or barium
sulphate
Whipworm infestation
Viral infections
Adverse effect of drugs and medications
What is the differential diagnosis?
- Several other skin conditions
may appear similar to erythema dyschromicum perstans because
they also result in discoloured skin patches.
- Lichen planus pigmentosus
(erythema dyschromicum perstans may be a variant of this disorder)
Multiple lesions of fixed drug eruption
Postinflammatory hyperpigmentation
Urticaria pigmentosa
Incontinentia pigmenti
Pinta
Leprosy
How is it diagnosed?
- In some cases the clinical
picture may be classical enough to diagnose the condition. A
skin biopsy may need to be performed in other cases to aid diagnosis.
This may reveal minor vacuolar degeneration of the basal layer
in early lesions, and pigmentary incontinence with dermal melanophages
in more established patches.
- What is the treatment?
- Unfortunately erythema dyschromicum
perstans is rather resistant to currently available treatments.
It may persist unchanged for years although some cases eventually
clear up by themselves.
- Treatments that may help improve
the appearance include:
- Topical steroids
Exposure to ultraviolet radiation
Pigment lasers e.g., Q-switched ruby laser
Chemical peels
The most successful systemic treatment has been clofazimine.
Dapsone, griseofulvin, hydroxychloroquine, isoniazid and corticosteroids
have been used successfully in a few cases.
-
- Erythema elevatum
diutinum (EED)
-
- Erythema elevatum diutinum
(EED) is a rare type of necrotising vasculitis that is characterised
by red, purple, brown or yellow papules (raised spot), plaques,
or nodules, found on the backs of the hands, other extensor surfaces
overlying joints, and on the buttocks.
- Erythema elevatum diutinum
Erythema elevatum diutinumErythema elevatum diutinumErythema
elevatum diutinum
Who gets erythema elevatum diutinum?
- EED may occur in any age group,
but patients are typically between 30 and 60 years old. It occurs
equally in men and women.
- What is the cause of erythema
elevatum diutinum?
- EED is classified as a small
vessel vasculitis. The cause of EED is not yet defined, but it
has been associated with the following conditions:
- Granuloma faciale
Recurrent bacterial infections (especially streptococci)
Viral infections (including hepatitis B and HIV)
Haematological diseases
Rheumatological diseases
What are the clinical features of erythema elevatum diutinum?
- Lesions usually start as papules
or nodules on the backs of the hands.
Other extensor surfaces affected include the knees, elbows, wrists,
ankles, fingers and toes. Buttocks, trunk, forearms, legs, palms
and soles may also be affected.
Lesions occurring on the face are indistinguishable from granuloma
faciale.
Lesions usually appear symmetrically.
Colour of lesions progress over time from yellow or pinkish to
red, purple or brown.
Lesions may enlarge during the day and go back to original size
overnight.
Rarely, blisters and ulcers may form.
Lesions usually feel firm and freely movable over the underlying
tissue.
EED can be symptomless or painful, or cause an itching or burning
sensation.
Symptoms can worsen after exposure to cold.
Arthralgia may be present.
How is erythema elevatum diutinum diagnosed?
- Several tests are available
to establish a diagnosis of EED.
- Skin biopsy (most important);
it shows leukocytoclastic vasculitis
Direct immunofluorescence
What is the treatment for erythema elevatum diutinum?
- EED is a chronic and progressive
skin disease that may last as long as 25 years. However, in some
cases after evolving over a 5-10 year period it may spontaneously
clear.
- Medication can be used to
limit progression of the disease.
- Dapsone is considered the
drug of choice for EED, mainly because of its rapid onset of
action and clinical experience has shown good responses. However,
lesions promptly recur following withdrawal of the drug.
- Other drugs that have been
occasionally reported to be effective include:
- Niacinamide
Colchicine
Hydroxychloroquine
Clofazimine
Cyclophosphamide.
Oral corticosteroids are generally ineffective.
- What is the outcome for erythema
elevatum diutinum?
- EED generally persists for
months, years or decades. It often recurs after apparently successful
treatment.
-
-
- Erythema induratum
(Bazin disease)
- Erythema induratum (Bazin
disease) presents as recurring nodules or lumps on the back of
the legs (mostly women) that may ulcerate and scar. It is a type
of nodular vasculitis
-
- Erythema infectiosum
-
- Erythema infectiosum is a
common childhood infection causing a slapped cheek appearance
and a rash. It is also known as fifth disease and human erythrovirus
infection.
- What is the cause of erythema
infectiosum?
- Erythema infectiosum is caused
by an erythrovirus, EVB19 or Parvovirus B19. It is a single-stranded
DNA virus that targets red cells in the bone marrow. It spreads
via respiratory droplets, and has an incubation period of 7–10
days.
- Who gets erythema infectiosum?
- Erythema infectiosum most
commonly affects young children and often occurs in several members
of the family or school class. Thirty percent of infected individuals
have no symptoms. It can also affect adults that have not been
previously exposed to the virus.
- Erythema infectiosum
Fifth diseaseFifth diseaseFifth disease
More images of erythema infectiosum ...
- What are the symptoms of erythema
infectiosum?
- Parvovirus B19 infection causes
nonspecific viral symptoms such as mild fever and headache at
first. The rash, erythema infectiosum, appears a few days later
with firm red cheeks, which feel burning hot. This lasts 2 to
4 days, and is followed by a pink rash on the limbs and sometimes
the trunk. This develops a lace-like or network pattern.
- Although most prominent in
the first few days, the rash can persist for up to six weeks
at least intermittently, and is most obvious when warm.
- Complications of erythema
infectiosum
- Although usually a mild childhood
condition, erythrovirus B19 infection can result in complications.
These include:
- Polyarthropathy in infected
adults (painful, swollen joints)
Aplastic crisis or potentially dangerous low blood cell count
in patients with haemolytic blood disorders such as autoimmune
haemolytic anaemia and sickle cell disease
Spontaneous abortion, intrauterine death (9%) or hydrops fetalis
in 3% of the offspring of infected pregnant women. This can occur
if erythema infectiosum occurs in the first half of pregnancy.
Parvovirus B19 does not cause congenital malformations. As the
risk of an adverse outcome is low, the infection is not routinely
screened for in pregnancy
Chronic parvovirus infection in immunodeficient patients, such
as organ transplant recipients, causing erythropoietin-resistant
anaemia, proteinuria, and glomerulosclerosis in a renal allograft
Rarely, encephalitis, hepatitis, non-occlusive bowel infarction,
amegakaryocytic thrombocytopenia, myositis and heart disease
How is the diagnosis of erythema infectiosum made?
- In most cases, erythema infectiosum
is a clinical diagnosis in a child with characteristic slapped
cheek and lacy rash. Parvovirus can cause other rashes such as
a papular purpuric gloves and socks syndrome. The diagnosis can
be confirmed by blood tests.
- Parvovirus serology: IgG,
IgM. This test is reported in about 7 days.
Parvovirus PCR is more sensitive. This test is reported in about
3 days.
In situ hybridisation or immunohistochemistry on biopsy specimens
If the child is unwell, or has haemolytic anaemia, a full blood
count should be performed. Ultrasound examination and Doppler
examination of at-risk pregancies can detect hydrops fetalis.
- Treatment of erythema infectiosum
- Erythema infectiosum is not
generally a serious condition. There is no specific treatment.
Affected children may remain at school, as the infectious stage
or viraemia occurs before the rash is evident.
- The application of an ice-cold
flannel can relieve the discomfort of burning hot cheeks.
Red blood cell transfusions and immunoglobulin therapy can be
successful in chronic parvovirus infection or during an aplastic
crisis.
Hydrops fetalis due to parvovirus infection is treated by intrauterine
transfusion.
ERYTHEMA MULTIFORME
Erythema multiforme is a hypersensitivity
reaction to infections or drugs or othetr trriggers characterised
by “target” lesions that are symmetrically distributed
with a propensity for the distal extremities. in the past SJS
and TEN were considered as the a severe form, erythema multiforme,
with its minimal mucous membrane involvement and less than 10
percent epidermal detachment, is now considered as a separate
entity.
Consensus classification
According to a consensus definition, Steven-Johnson syndrome was
separated from the erythema multiforme spectrum and added to toxic
epidermal necrolysis. [3] Essentially Steven-Johnson syndrome
and toxic epidermal necrolysis (TEN) are considered severity variants
of a single entity. The 2 spectra are now divided into the following:
(1) erythema multiforme consisting of erythema minor and major
and (2) Steven-Johnson syndrome / toxic epidermal necrolysis (SJS/TEN).
The clinical descriptions are as follows:
Erythema multiforme minor - Typical targets or raised, edematous
papules distributed acrally
Erythema multiforme major - Typical targets or raised, edematous
papules distributed acrally with involvement of one or more mucous
membranes; epidermal detachment involves less than 10% of total
body surface area (TBSA).
SJS/TEN - Widespread blisters predominant on the trunk and face,
presenting with erythematous or pruritic macules and one or more
mucous membrane erosions; epidermal detachment is less than 10%
TBSA for Steven-Johnson syndrome / toxic epidermal necrolysis
and 30% or more for toxic epidermal necrolysis.
May affect any age group but
is most common in young adults (20–40 years of age).
Causes
- Allergic reaction to drugs
such as penicillin, sulphonamides and barbiturates.
- Infections such as Mycoplasma
pneumoniae, herpes
simplex virus (cold sore virus) and fungal ifnfections. Erythema
multiforme can be recurrent, with multiple episodes per year
for many years. This is believed to be nearly always due to HSV-1
infection.
- Medication including barbiturates,
non-steroidal anti-inflammatory drugs, penicillins, sulphonamides,
phenothiazines and anticonvulsants.antibiotics, such as sulfonamides,
tetracyclines, amoxicillin and ampicillin
non-steroidal anti-inflammatory drugs (NSAIDs), such as ibuprofen
anticonvulsants (used to treat epilepsy), such as phenytoin and
barbiturates
- Internal disease.
- Pregnancy.
- Unknown.
- There is a genetic tendency
to erythema multiforme. Certain tissue types are more often found
in people with herpes-associated erythema multiforme (HLA-DQw3)
and recurrent erythema multiforme (HLA-B15, -B35, -A33, -DR53,
-DQB1*0301).
- Cell-mediated immunity appears
to be responsible for the destruction of epithelial cells. Early
in the disease process, the epidermis becomes infiltrated with
CD8 T lymphocytes and macrophages, whereas the dermis displays
a slight influx of CD4 lymphocytes. These immunologically active
cells are not present in sufficient numbers to be directly responsible
for epithelial cell death. Instead, they release diffusable cytokines,
which mediate the inflammatory reaction and resultant apoptosis
of epithelial cells. In some patients, circulating T cells transiently
demonstrate (for < 30 d) a T-helper cell type 1 (TH1) cytokine
response (interferon [IFN] gamma, tumor necrosis factor [TNF]
alpha, interleukin [IL] 2). Results of immunohistochemical analysis
have also shown lesion blister fluid to contain TNF, an important
proinflammatory cytokine.
- In addition, there have been
reports of erythema multiforme associated with vaccines (diphtheria-tetanus,13
hepatitis B,14 smallpox15), other viruses (varicella zoster virus,16
hepatitis C,17 cytomagalovirus,18,19 and human immunodeficiency
virus20), and some newer medications (candesartan cilexetil [Atacand],21
rofecoxib [Vioxx; withdrawn from the U.S. market],22 metformin
[Glucophage],23 adalimumab [Humira],24 bupropion [Well-butrin],25
and ciprofloxacin [Cipro]26).
- Symptoms
- Characteristic lesion is a
"Target" or "bull's eye" which has a regular
round shape and three concentric zones: a central dusky or darker
red area, a paler pink or edematous zone, and a peripheral red
ring. Atypical target lesions have only two zones, the dusky
or darker red center and a pink or lighter red border.
- Patients may experience itching
and burning at the site of the eruption
- May be preceded by a cough,
fever, sorethroat, malaise (feeling of illness) or headache.
- Mucosal lesions, may occur,
typically develop a few days after the skin rash begins but are
limited only to the oral cavity. They may consist initially of
redness of the lips and inside cheek. Sometimes blisters develop
and quickly break to form erosions and ulcers.
- In erythema multiforme minor,
mucous membrane involvement is absent or mild. Mucosal changes,
if present,
- In
severe cases, the mucous membranes (eyes, mouth, nasal passages
and genitals) may be ulcerated. This form is called Steven's
Johnson's syndrome. Another more severe form is called toxic
epidermal necrolysis. In this form the, the rashes progress
rapidly to become blisters that slough off leaving raw areas
like a scald.
Mucosal lesions may occur but usually are limited to the oral
cavity.1 Erythema multiforme resolves spontaneously in three
to five weeks without sequelae, but it may recur.3 Patients in
whom it recurs may have multiple episodes per year. In a study
involving 65 patients with recurrent erythema multiforme, the
mean number of attacks per year was six, with a range of two
to 24; the mean duration of the disease was 9.5 years.
 |
Erythema multiforme.
Click of
image for larger view |
Complications
- Secondary infection of damaged
skin.
-
- What you can do
- You should consult a doctor.Most
people with erythema multiforme make a full recovery within a
few weeks. There aren't usually any further problems and the
skin normally heals without scarring
-
- What the doctor may do
- Erythema multiforme is a clinical
diagnosis, although skin biopsy may be required to exclude other
conditions. The histology of erythema multiforme is characteristic
but not diagnostic. It varies with the age of the lesion, its
appearance, and which part is biopsied.
- Other tests may be done looking
for infections commonly seen in association with erythema multiforme,
such as mycoplasma.
- For the majority of cases,
no treatment is required, as the rash settles by itself over
several weeks without complications.
- Mild cases of erythema multiforme do not require treatment.6
Oral antihistamines and topical steroids may
be used to provide symptom relief.
- Prescribe systemic steroids
(oral or intravenous) Prednisone may be used in patients with
many lesions
- Supportive/symptomatic treatment
may be necessary.
- Itch — oral antihistamines
and/or topical corticosteroids may help.
Oral pain — mouthwashes containing local anaesthetic and
antiseptic reduce pain and secondary infection.
Eye involvement should be assessed and treated by an ophthalmologist.
Erythema multiforme major may require hospital admission for
supportive care, particularly if severe oral involvement restricts
drinking.This can be severe and require hospitalisation
due to difficulty eating and drinking. Whether this is a limited
form of erythema multiforme has not been determined.
The role of oral corticosteroids remains controversial, as no
controlled studies have shown any benefit. However for severe
disease 0.5–1 mg/kg/d prednis(ol)one is often used early
in the disease process.
- Treat the underlying cause.
- TOP
Erythema nodosum is a type of
panniculitis (inflammation of the fat) characterised by painful
inflamed nodules (large swellings) on the limbs, most commonly
on the shins.
Generally, it is idiopathic,
although the most common identifiable cause is streptococcal pharyngitis.
Erythema nodosum may be the first sign of a systemic disease such
as tuberculosis, bacterial or deep fungal infection, sarcoidosis,
inflammatory bowel disease, or cancer. Certain drugs, including
oral contraceptives and some antibiotics, also may be etiologic.
The hallmark of erythema nodosum is tender, erythematous, subcutaneous
nodules that typically are located symmetrically on the anterior
surface of the lower extremities. Erythema nodosum does not ulcerate
and usually resolves without atrophy or scarring. Most direct
and indirect evidence supports the involvement of a type IV delayed
hypersensitivity response to numerous antigens. A deep incisional
or excisional biopsy specimen should be obtained for adequate
visualization. Erythema nodosum represents an inflammatory process
involving the septa between subcutaneous fat lobules, with an
absence of vasculitis and the presence of radial granulomas. Diagnostic
evaluation after comprehensive history and physical examination
includes complete blood count with differential; erythrocyte sedimentation
rate, C-reactive protein level, or both; testing for streptococcal
infection (i.e., throat culture, rapid antigen test, antistreptolysin-O
titer, and polymerase chain reaction assay); and biopsy. Patients
should be stratified by risk for tuberculosis. Further evaluation
(e.g., purified protein derivative test, chest radiography, stool
cultures) varies based on the individual. Erythema nodosum tends
to be self-limited. Any underlying disorders should be treated
and supportive care provided. Pain can be managed with nonsteroidal
anti-inflammatory drugs.
- Cause
- Erythema nodosum appears
to be a hypersensitivity reaction to different causes.
- Infections: streptococcal
pharyngitis (28 to 48 percent), Yersinia spp. (in Europe), mycoplasma
pneumoniae, chlamydia, histoplasmosis, coccidioidomycosis, mycobacteria,
virus
- Sarcoidosis; (11 to 25
percent) with enlargement of the lymph nodes (bihilar lymphadenopathy)
in the lungs.
Tuberculosis (TB); erythema nodosum occurs with the primary infection
with TB. TB in New Zealand is currently uncommon.
- Drugs (3 to 10 percent), eg.,
due to sulphur drugs, iodides, penicillin and orall contraceptives
Pregnancy (2 to 5 percent) - EN may occur in pregnancy, clear
after delivery, then recur in subsequent pregnancies.
Inflammatory bowel disease (ulcerative colitis or Crohn disease)
Idiopathic (up to 55 percent)
- Behcets
Erythema nodosum leprosum is a particular variant of erythema
nodosum that affects some people being treated for leprosy.
-
- Symptoms
- Red painful swellings which
later become purple then assume a bruise-like appearance as they
fade.
- Usually occur on the shins,
sometimes on the thighs and outer aspects of the arms.
- May be associated with fever,
malaise (feeling of illness), headache, muscle and joint pains.
- Subside after 3 - 6 weeks.
- Sometimes, erythema nodosum
may become a chronic or persistent disorder lasting for 6 months
and occasionally for years.
- Complications
- Depends on the underlying
cause.
-
- What you can do
- You should consult a doctor.
-
- What the doctor may do
- Determine and treat the underlying
cause.
- Tests done in patients with
erythema nodosum include:
- Throat swab
Sputum or gastric washing if TB is suspected
Complete blood count and C-reactive protein (CRP) and/or erythrocyte
sedimentation rate (ESR)
ASO titre (a test for streptococcal infection)
Chest X-ray (looking for TB and sarcoidosis)
Virus studies
Yersinia titres
Mantoux test or QuantiFERON gold (tests for TB)
- Complete blood count with
differential; erythrocyte sedimentation rate and C-reactive protein
levels
Evaluation for streptoccocal infection (i.e., throat culture
for group A streptococci, rapid antigen test, antistreptolysin-O
titer, and polymerase chain reaction assay)
Excisional biopsy (when clinical diagnosis is in doubt); key
histologic findings are septal panniculitis, lymphocytic infiltrate
with neutrophils, actinic (Miescher's) radial granulomas, absence
of vasculitis, and no organisms
Clinical suspicion of chronic disease (e.g., sarcoidosis, tuberculosis);
purified protein derivative test, chest radiography
Stool culture and evaluation for ova and parasites in patients
with diarrhea or gastrointestinal symptoms; consider evaluation
for inflammatory bowel disease
- Bed rest.
- Prescribe painkillers or nonsteroidal
antiinflammatory drugs (NSAIDs), potassium iodide, oral tetracyclines
or oral steroids.
- Although erythema nodosum
can be exquisitely tender, it tends to be self-limited.
Treatment of erythema nodosum
- If an underlying infection
is found, this should be treated.
Bed rest is advised if pain and/or swelling is severe.
Firm supportive bandages or light compression stockings should
be worn.
Anti-inflammatory medications reduce discomfort
Potassium iodide has been reported to be effective but is not
easy to obtain.
Oral tetracyclines have anti-inflammatory properties and may
reduce discomfort and duration of disease.
- TOP
-
- Erythema marginatum
rheumaticum
-
- Erythema marginatum rheumaticum
– This is a characteristic type of annular erythema that
occurs in about 10% of first attacks of ARF in children; it is
very rare in adults. The rash can be difficult to detect in dark-skinned
people. When present, it is found on the trunk and upper arms
and legs, but almost never on the face, palms or soles. The rash
appears as pink or red macules (flat spots) or papules (small
lumps), which spread outwards in a circular shape. As the lesions
advance, the edges become raised and red, and the centre clears.
The lesions are not itchy or painful, and sometimes go unnoticed
by the patient. The lesions can fade and reappear within hours,
reappearing in hot conditions. Erythema marginatum may persist
intermittently for weeks to months, even after successful treatment
of ARF.
-
- Erythema toxicum neonatorum
-
- Erythema toxicum neonatorum
is common and benigh eruption in newborns, affecting more than
50% of full term babies. It appears to be less common in premature
babies. Erythema toxicum neonatorum may be present at birth but
more often appears during the second or third day of life.
-
- Typical lesions consist of
erythematous, 2- to 3-mm macules and papules that evolve into
pustules. Each pustule is surrounded by a blotchy area of erythema,
leading to what is classically described as a “flea-bitten”
appearance. Lesions usually occur on the face, trunk, and proximal
extremities. Palms and soles are not involved.
- Diagnosis
- The diagnosis of toxic erythema
of the newborn is usually made on clinical grounds.
- Although toxic erythema of
the newborn is benign and requires no treatment, a number of
differential diagnoses should be considered.
ERYTHRASMA
Erythrasma is a superficial
bacterial infection of the body folds such as the toe webs, groin
and armpits. It is more common in warm climates and usually affects
young adults. Widespread infections are associated with diabetes
mellitus.
- Cause
- Infection with a bacteria
called Corynebacterium minutissimum.
Aggravating factors
- Diabetes
- A warm, humid climate.
Symptoms
Erythrasma is classified into
3 types according to location.
Interdigital erythrasma: between
3rd, 4th and 5th toe web spaces
Intertriginous erythrasma: in armpits, groin, under the breasts
and umbilicus
Generalised/disciform erythrasma: on the trunk
-
- Well-defined pink or brown
dry patches with fine scales and wrinkling in the armpits, groins
and buttock cleft. They start off pink or red and becomes brown
and scaly late.
- Common sites for erythrasma
are armpits, groin and between the toes. Intergluteal fold, submammary,
and periumbilical skin may also be affected.
- Scaling
and maceration of the toe webs.
- Mild itching may be present.
-
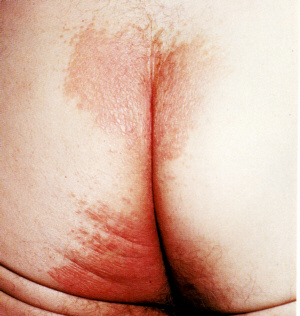 |
Erythrasma
Click on
image for larger view |
- Erythrasma may coexist with
or be confused with other causes of intertrigo (rashes in the
skin folds) including fungal infections such as tinea or Candida
albicans (thrush).
-
- What you can do
- You should consult a doctor.
- keeping the area clean and
dry
- Wear loose clothes.Aluminum
chloride solution to inhibit sweating and moisture
-
- What the doctor may do
- Confirm the diagnosis..
- The affected areas fluoresce
a coral red colour coproporphyrin III when illuminated with a
Wood's lamp. Another way to diagnose erythrasma is by skin scrapings:
microscopy may reveal gram-positive filamentous rods. Methylene
blue also stains C minutissimum. The coral-red fluorescence of
scales seen under Wood light is secondary to the production of
porphyrin by these diphtheroids.
- Topical antibiotic lotions
such as erythromycin or clindamycin. Fusidic acid cream.
Whitfield's ointment (a mixture of benzoic acid and salicylic
acid)
- For a more aggressive treatment,
oral antibiotics such as erythromycin or clarithromycin can also
be used
- Relapses are common.
- Photodynamic therapy using
red light (broadband, peak at 635 nm) has also been used to treat
patients with erythrasma.Oral antibiotics such as erythromycin
or clarithromycin
- TOP
ERYTHRODERMA AND EXFOLIATIVE DERMATITIS
Erythroderma refers to a diffuse
reddish inflammation of the skin affecting more than 90% of the
body surface. The term exfoliative dermatitis is used when most
of the skin is exfoliating or shedding.
The extent of the skin changes
can obscure the primary lesion making it difficult to diagnose
the underlying cause.
Erythroderma is the clinical
presentation of a wide range of cutaneous and systemic diseases
(including psoriasis and atopic dermatitis), drug hypersensitivity
reactions, and more rarely Sézary syndrome, a leukemic
subtype of cutaneous T-cell lymphoma. Although uncommon in pediatric
patients, erythroderma may be the clinical presentation of a wide
range of acquired and inherited diseases, including infections,
inflammatory skin diseases, ichthyoses, and congenital immunodeficiencies.
The mechanism of erythroderma
is unknown. Adhesion molecules and their ligands (vascular cell
adhesion molecule 1, intercellular adhesion molecule 1, E-selectin
and P-selectin) play a major role in endothelial-leucocyte interaction,
which affect the binding, transmigration and infiltration of lymphocytes
and mononuclear cells during inflammation, injury or immunologic
reaction. The rise in adhesion molecule expression stimulates
dermal inflammation, which leads to epidermal proliferation and
increased production of inflammatory mediators. However, there
is no difference in adhesion molecule expression on endothelial
cells between different types of erythroderma.Sézary Syndrome
is a Th2 disorder with a selective expression of CCR4, whereas
inflammatory erythroderma shares an overexpression of both Th1-
and Th2- related chemokine receptors, suggesting a different pathophysiologic
mechanism.
Spread of a pre-existing
skin disease such as atopic
dermatitis, seborrhoeic
dermatitis, varicose eczema,
contact dermatitis, psoriasis, pityriasis
rubra pilaris and pemphigus
foliaceus and bullous pemphigoid, Papuloerythroderma of Ofuji,
norwegian crusted scabies.
Drug
hypersensitivity reactions (see drug
eruption) - especially antimalarials, allopurinol (gout medicine),
non-steroidal anti-inflammatory drugs, phenytoin (used to treat
epilepsy), gold (used to treat rheumatoid arthritis) and penicillin
and sulphonamide antibiotics. Underlying cancer - Sézary syndrome
(a leukemic subtype of cutaneous T-cell lymphoma), lymphoma and
leukaemia. Several
very rare congenital ichthyotic conditions and congenital immunodeficiencies.
Genetic testing could be beneficial for certain congenital erythrodermas
such as Netherton's syndrome and Conradi-Hunermann syndrome. Erythroderma
may be the first clinical manifestation of an immunodeficiency
syndrome, especially in the presence of diarrhea and profound
failure to thrive. Immunodeficiency screening must be considered
in these cases. Graft-versus-host
disease HIV infection
Idiopathic
Unknown.
Symptoms
- Symptoms
Diffuse red inflamed skin.
Variable
degrees of scaling and shedding (exfoliation) as the condition
progresses. Itching.
Lymphadenopathy
is often present but is more often reactive than malignant. In
difficult cases, a lymph node biopsy is recommended. However,
the pathologist must be informed that the patient is erythrodermic
for a reliable histopathological interpretation to be made.Lymph
nodes become swollen (generalised dermatopathic lymphadenopathy).
Fatigue.
Fever.
Shivering
from excessive heat loss. Failure of the mechanical barrier will
result in loss of normal temperature control with failure to maintain
the core body temperature.
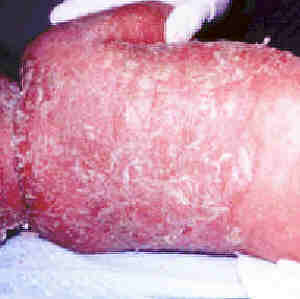 |
Erythroderma.
Click on
image for larger view |
Clues may be present as to the underlying cause. Serous
ooze, resulting in clothes and dressings sticking to the skin
and an unpleasant smell, is characteristic of atopic erythroderma.
Persistence of circumscribed scaly plaques in certain sites such
as elbows and knees suggests psoriasis.
Islands of sparing, follicular prominence, orange-hue to keratoderma
are typical of pityriasis rubra pilaris.
Subungual hyperkeratosis, crusting on palms and soles, and burrows
are indicative of crusted scabies.
Sparing of abdominal creases (deck chair sign) is typical of papuloerythroderma
of Ofuji. Complications
Hypothermia
(low body temperature) due to increased heat loss from the dilated
blood vessels in the skin.Shivering and hypothermia may occur
even though the skin feels deceptively warm. Cardiac failure due to increased blood
flow through the skin putting a strain on the heart.Persistent
inflammation of the skin leads to marked peripheral vasodilation
and increased cutaneous blood flow. In combination with increased
percutaneous loss of fluid, these can lead to high-output cardiac
failure, especially in elderly patients. Hypoalbuminemia
(low serum albumin) is common and probably has several causes
including increased plasma volume, decreased synthesis, increased
metabolism, and protein loss via scaling and exudation.
Telogen
effluvium and onychodystrophy may develop when erythroderma has
been present for some weeks. Iron
deficiency anaemia. Weight loss.
Hypoalbuminemia is common and probably has several causes including
increased plasma volume, decreased synthesis, increased metabolism,
and protein loss via scaling and exudation. Peripheral edema is common and can be due to a
variety of causes, such as a shift of fluid into extracellular
spaces, hypoalbuminaemia, concomitant cardiac failure and inflammation
resulting from the primary skin disease. Occasionally, the increased
permeability is severe enough to justify plasma infusions and
parental steroids. Severe pulmonary
edema secondary to capillary leak syndrome and adult respiratory
distress syndrome (ARDS) is a complication of erythroderma.
Colonization of
the skin by Stahylococcus aureus is common and can lead to secondary
cutaneous infections. Respiratory infections including pneumonia
are common.
- An eruption of warty papules resembling seborrheic keratoses,
"Murray Williams warts" can occasionally occur following
resolution of an inflammatory dermatosis.Longstanding erythroderma
may result in pigmentary changes (brown and/or white skin patches).
-
- What you can do
You should consult a doctor.
-
- What the doctor may do
Hospitalise for management.Investigation
should involve hematology, urea and electrolytes, liver function
tests and blood culture. Elevated erythrocyte sedimentation rate,
raised IgE, anemia, hypoalbuminemia and hyperglobulinemia are
frequent findings. Eosinophilia is generally a nonspecific finding,
although a highly elevated count may be associated with malignancy,
mainly T-cell lymphoma.
Skin biopsies from several sites may be taken if the cause is
unknown. They tend to show nonspecific inflammation on histopathology.
Diagnostic features may be present however. Direct immunofluorescence is of benefit if an
autoimmune blistering disease or connective tissue disease is
considered. Determine and treat
the underlying cause. Prescribe oral
and or topical steroids.
Prescribe
antihistamines to
reduce itching.
- Treat the complications.
The course of idiopathic erythroderma is unpredictable. It may
persist for a long time with periods of acute exacerbation.
-
- TOP
penile intraepithelial neoplasia
What is penile intraepithelial neoplasia? Penile intraepithelial
neoplasia is a rare pre-cancerous disease of the outer skin layer
(epidermis) of the penis. Other names for penile intraepithelial
neoplasia include: Squamous intraepithelial lesion
Erythroplasia of Queyrat
Bowen disease of the penis
in-situ squamous cell carcinoma of the penis
P.I.N.
How is penile intraepithelial neoplasia recognised? The diagnosis
is often delayed, because penile intraepithelial neoplasia may
resemble other conditions such as balanitis, candidiasis, dermatitis
and psoriasis. Lesions are single or multiple, red plaques
on the glans or inner aspect of the foreskin. They may have a
smooth, velvety, moist, scaly, eroded or warty surface. The following
signs and symptoms may occur: Redness and inflammation
Itching
Crusting or scaling
Pain
Ulcers
Bleeding
In the late stages, discharge from penis, difficulty pulling back
foreskin or difficulty passing urine
Images of penile intraepithelial neoplasia Who is at risk
of penile intraepithelial neoplasia and what causes it? Uncircumcised
males over 50 years of age are most at risk of getting penile
intraepithelial neoplasia, although it may rarely occur in younger
men. Penile intraepithelial neoplasia is associated with:
Chronic infection with human papilloma virus (HPV), the cause
of genital warts. HPV-16 is the most common type identified.
Chronic skin disease, especially lichen sclerosus and lichen planus
Smoking
Immune suppression by medications or disease
Chronic irritation by urine, friction or injury to the penile
area.
If left untreated, 10–30% of cases develop into invasive
squamous cell carcinoma (cancer) of the penis. What is the
treatment for penile intraepithelial neoplasia? Skin biopsy
should be performed to confirm the diagnosis, as it may resemble
other forms of chronic balanitis. Biopsy is also essential to
rule out invasive squamous cell carcinoma, which requires more
aggressive treatment. It is important to maintain good genital
hygiene. Penile intraepithelial neoplasia can be treated in several
different ways. Multidisciplinary care may be necessary. 5-fluorouracil
cream
Imiquimod cream
Cryotherapy
Curettage and cautery
Laser vaporisation
Photodynamic therapy
Radiotherapy
Excision
Interferon alpha
Mohs micrographic surgery appears to be highly effective and the
surgical treatment of choice in severe or recurrent cases of penile
intraepithelial neoplasia. The disease recurs in 3-10% of
patients, so close follow-up is necessary to ensure a complete
cure. Partners of patients with penile intraepithelial neoplasia
should be screened for other forms of intraepithelial neoplasia
caused by human papilloma virus in the genital area (cervical,
vulvar and anal cancer). Many national immunisation programmes
now include a vaccine against the causative human papillomaviruses
HPV-16 and 18. Vaccination of boys and young men should be included,
to reduce the risk of developing penile intraepithelial cancer
in the future.
extramammary Paget disease
What is extramammary Paget disease? Extramammary Paget
disease is an uncommon cancer characterised by a chronic eczema-like
rash of the skin around the anogenital regions of males and females.
Under the microscope extramammary Paget disease looks very similar
to the more common type of mammary Paget disease that occurs on
the breast. Who gets extramammary Paget disease? Extramammary
Paget disease most commonly occurs in the vulva of women aged
between 50–60 years. It can also affect males of similar
age. How is extramammary Paget disease classified? Extramammary
Paget disease has been classified into several subtypes. Type
1a primary cutaneous extramammary Paget disease arises from apocrine
glands within the epidermis (in situ) or underlying skin appendages
Type 1b primary cutaneous extramammary Paget disease (15-25%)
is associated with invasive Paget disease or adenocarcinoma in
situ.
Type 2 extramammary Paget disease originates from underlying anal
or rectal adenocarcinoma
Type 3 extramammary Paget disease originates from bladder adenocarcinoma
Sometimes the extramammary Paget disease has been present for
10–15 years before evidence of cancer or metastases appear.
What are the clinical features of extramammary Paget disease?
The most common symptom of extramammary Paget disease is mild
to intense itching of a lesion found around the groin, genitalia,
perineum or perianal area. Pain and bleeding may occur from scratching
lesions that have been around for a long time. Thickened plaques
may form that can become red, scaly and crusty. Although they
may appear similar to eczema, they fail to clear up with topical
steroid creams. Extramammary Paget disease
Extramammary Paget diseaseExtramammary Paget diseaseExtramammary
Paget disease
What sites are affected? In women the most common area involved
is the vulva. First symptoms are usually itching and burning of
of one or more persistent plaques. These may spread to the labia,
mons pubis, vagina and thighs. Perianal lesions may extend up
into the anal canal. The location of extramammary Paget disease
is useful in predicting the risk of associated cancer. For example,
25-35% of extramammary Paget disease arising near the anus is
associated with an underlying colorectal cancer. How is extramammary
Paget disease diagnosed? Skin biopsy of the lesion is performed
to get an accurate diagnosis of extramammary Paget disease as
there are several other genital skin diseases that may appear
similar. Under microscopy, the presence of Paget cells along with
other histological findings confirms diagnosis. Special stains
may be necessary to distinguish Paget disease from early melanoma
(melanoma in situ). See extramammary Paget disease pathology.
Further tests may include: Evaluation of lymph nodes by
ultrasound scan or fine needle aspirate
Search for other malignancies including PAP smear (cervical smear),
pelvic imaging to look for underlying cancer, colonoscopy, mammography.
What is the treatment of extramammary Paget disease? Wide
local excision, vulvectomy, or if available, margin-controlled
surgical excision (Mohs micrographic surgery) is the standard
treatment for extramammary Paget disease . Paraffin sections are
preferred over frozen sections. The margin is sometimes difficult
to define particularly when lesions are spread sporadically throughout
the anogenital region. Reconstruction may require skin grafting
or flap repair. Recurrence is common (30-50%), so patients
should be re-examined every 3 months after surgery for the next
2 years, after which annual follow-ups are recommended. Recurrence
generally leads to further surgery. Non-surgical treatments
for recurrent disease may include: Radiotherapy
Laser ablation
Photodynamic therapy
5-fluorouracil cream
Imiquimod cream
EYE
BAGS
Eye bags are a common cause
of concern because they make a person look old and less alert.
- Cause
- Herniation of fat through
a weakening in the orbicularis oculi muscle.
- Loss of skin elasticity due
to ageing.
- Swelling due to fluid accumulating
as a result of lack of sleep, stress, allergy or illness.
- Inherited condition known
as blepharochalasia.
Symptoms
- Loose skin under the eyes.
- Swelling under the eyes.
What you can do
- The swelling, if due to fluid
aculating, can be reduced by using a cool compress
or cucumber slices.
What the doctor can do
- Exclude allergies and other
illnesses.
- Perform blepharoplasty (eyelid
lift).
-
- TOP